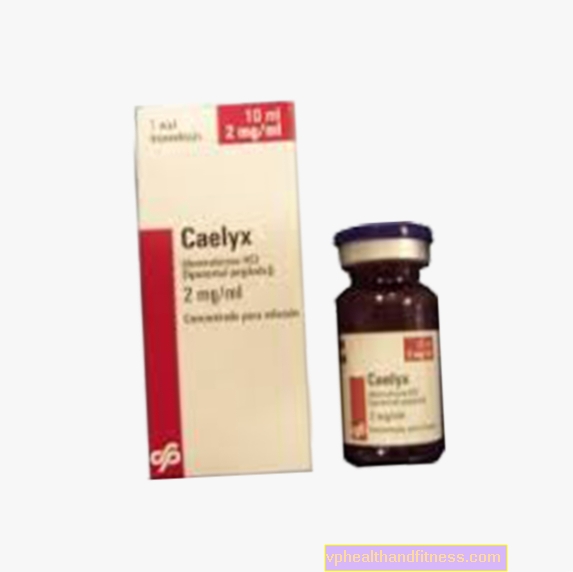
1 ml de preparat conține 2 mg clorhidrat de doxorubicină în lipozomi pegilați. Preparatul conține zaharoză și fosfatidilcolină din soia complet hidrogenată (din soia).
| Nume | Conținutul pachetului | Substanța activă | Preț 100% | Modificat ultima dată |
| Caelyx | 1 flacon, final de făcut soluţie a inf. | Clorhidrat de doxorubicină | 2019-04-05 |
Acțiune
Antibiotic citotoxic antraciclinic obținut din culturi de Streptomyces peucetius var. caesius. Medicamentul se acumulează între perechi de baze adiacente în dubla helix ADN, împiedicându-l să se desfășoare necesar pentru replicare. Acest lucru duce la o inhibare a sintezei ADN, ARN și proteine. Forma lipozomală pegilată a clorhidratului de doxorubicină prelungește timpul de ședere al medicamentului în sistemul circulator. Farmacocinetica preparatului diferă semnificativ de formele standard de doxorubicină. La doze mai mici (10 mg / m2 - 20 mg / m2) preparatul prezintă farmacocinetică liniară; în doza de 10 mg / m2. - 60 mg / m2 farmacocinetica este neliniară. Preparatul, spre deosebire de formele standard, care sunt distribuite în mare parte în țesuturi, rămâne în principal în volumul lichidului vascular, iar eliminarea doxorubicinei din sânge este dependentă de purtătorul lipozomal. Doxorubicina devine disponibilă după ce lipozomii părăsesc vasul și intră în compartimentul tisular. După administrarea de doze echivalente de preparat și forme standard, concentrația sanguină și valorile ASC ale formei lipozomale pegilate sunt mai mari decât cele obținute cu formele standard de clorhidrat de doxorubicină. T0.5 este de 24-231 h, cu o medie de 73,9 h.
Dozare
Intravenos, prin infuzie. Administrați numai sub supravegherea unui medic oncolog specialist cu experiență în utilizarea medicamentelor citotoxice. Preparatul nu poate fi utilizat interschimbabil cu alte forme farmaceutice de clorhidrat de doxorubicină. Cancer de sân sau cancer ovarian: 50 mg / m2 la fiecare 4 săptămâni, atâta timp cât boala nu progresează și atât timp cât pacientul tolerează tratamentul. Mielom multiplu: 30 mg / m2 în ziua 4 a unui ciclu de tratament cu bortezomib de 3 săptămâni sub formă de perfuzie de 1 oră imediat după perfuzia cu bortezomib. Regimul de tratament cu bortezomib este de 1,3 mg / m2 în zilele 1, 4, 8 și 11 pentru cicluri de tratament de 3 săptămâni. Tratamentul trebuie să continue atât timp cât un răspuns la tratament este menținut atât timp cât pacientul tolerează tratamentul. Ziua tratamentului combinat (ciclul de ziua 4) poate fi amânată la 48 de ore dacă este indicat din punct de vedere medical, dar intervalul dintre dozele consecutive de bortezomib nu trebuie să fie mai mic de 72 de ore Sarcomul SIDA Kaposi: 20 mg / m2. la fiecare 2-3 săptămâni. Pauzele mai scurte de 10 zile trebuie evitate, deoarece nu se pot exclude acumularea de medicamente și toxicitatea crescută. Se recomandă continuarea tratamentului timp de 2-3 luni. Tratamentul trebuie continuat după cum este necesar pentru a menține un răspuns terapeutic. Modificarea dozelor în caz de reacții adverse. Pentru a gestiona reacțiile adverse (cum ar fi roșeața palmară și a tălpilor - EPP, stomatită sau toxicitate hematologică), doza poate fi redusă sau administrată ulterior. Eritrodisestezia palmar-plantară (EIP). Primul. La 4 săptămâni după doza anterioară a preparatului - 100% din doză trebuie administrată dacă pacientul nu a prezentat nicio toxicitate cutanată anterioară a treia sau a patra și, dacă a apărut - așteptați încă o săptămână. Primul. toxicitate în a 5-a săptămână după doza anterioară a preparatului - 100% din doză trebuie administrată dacă pacientul nu a prezentat nicio toxicitate cutanată anterioară a 3-a sau a 4-a și dacă a apărut - așteptați o săptămână suplimentară pe 1. toxicitate în a 6-a săptămână după doza anterioară a preparatului - reduceți doza cu 25%; reveniți la pauza de 4 săptămâni. Al 2-lea. Toxicitate (eritem, descuamare sau umflături care interferează cu, dar nu împiedică activitatea fizică normală; vezicule mici sau ulcere cu diametru de Stomatită. Prima toxicitate (ulcerație nedureroasă, eritem sau ușoară durere) în a 4-a săptămână după doza anterioară a preparatului - ar trebui să fie administrați 100% din doză dacă pacientul nu a dezvoltat stomatită înainte de a 3-a sau a 4-a stomatită și dacă pacientul nu a avut o stomatită anterioară, așteptați încă o săptămână 1 de toxicitate în a 5-a săptămână după doza anterioară a preparatului - trebuie administrată 100% din doză, dacă nu stomatita anterioară a apărut în etapa a 3-a sau a 4-a și, dacă a apărut - așteptați o săptămână suplimentară a primei săptămâni de toxicitate în a 6-a săptămână după doza anterioară a preparatului - reduceți doza cu 25%; reveniți la pauza de 4 săptămâni sau, pe baza evaluării medicului, opriți administrarea A doua toxicitate (eritem dureros, umflare sau ulcerație, dar cu posibilitatea de a mânca) în a 4-a săptămână după doza anterioară a preparatului - așteptați încă o săptămână z. 2. toxicitate în a 5-a săptămână după doza anterioară a preparatului - așteptați încă o săptămână a doua. toxicitate în a 6-a săptămână după doza anterioară a preparatului - reduceți doza cu 25%; reveniți la pauza de 4 săptămâni sau opriți administrarea pe baza judecății medicului dumneavoastră. 3. Toxicitate (eritem dureros, umflare sau ulcerație fără posibilitatea de a mânca) în a 4-a săptămână după doza anterioară a preparatului - așteptați încă o săptămână. toxicitate în a 5-a săptămână după doza anterioară a preparatului - așteptați încă o săptămână. La 6 săptămâni după doza anterioară a preparatului - întrerupeți administrarea. A 4-a. toxicitate (nutriție parenterală sau enterală necesară) în a 4-a săptămână după doza anterioară a preparatului - așteptați încă o săptămână 4. toxicitate în a 5-a săptămână după doza anterioară a preparatului - așteptați încă o săptămână 4. La 6 săptămâni după doza anterioară a preparatului - întrerupeți administrarea. Programul de modificare a dozei de mai sus poate fi utilizat și la pacienții cu sarcom SIDA Kaposi și la pacienții cu mielom multiplu care primesc terapie combinată cu bortezomib. Efect toxic asupra sistemului hematopoietic (cancer mamar sau ovarian) - 1: ANC (număr absolut de neutrofile) 1500-1900 / mm3, trombocite 75.000-150.000 / mm3 - reluați tratamentul fără reducerea dozei; 2: ANC 1000 - 3, trombocite 50.000 - 3, așteptați până la ANC ≥ 1.500 / mm3 și trombocite ≥ 75.000 / mm3, re-administrați fără reducerea dozei; Etapa a treia: ANC 500 - 3, trombocite 25.000 - 3 - așteptați până la ANC ≥ 1.500 / mm3 și trombocite ≥ 75.000 / mm3, re-administrați fără reducerea dozei; Al patrulea: ANC 3, trombocite 3 - așteptați până când ANC ≥ 1.500 / mm3 și trombocite ≥75.000 / mm3, reduceți doza cu 25% sau continuați tratamentul cu doza completă prin administrarea factorului de creștere. Efect toxic asupra sistemului hematopoietic (pacienți cu sarcom Kaposi în cursul SIDA) - tratamentul cu preparatul trebuie întrerupt temporar când ANC este 3 și / sau numărul de trombocite este 3, simultan, pentru a crește numărul de celule sanguine atunci când ANC este 3, în ciclurile următoare Se poate administra G-CSF (sau GM-CSF). Modificarea dozei pentru pacienții cu mielom multiplu în timpul terapiei combinate cu bortezomib. Febra ≥38 ° C și ANC 3 - nu administrați doza adecvată de doxorubicină dacă simptomele au apărut înainte de a 4-a zi a ciclului de tratament; dacă după ziua 4, următoarea doză trebuie redusă cu 25%; reduceți următoarea doză de bortezomib cu 25%. În orice zi de administrare după prima zi a fiecărui ciclu: numărul de trombocite 3 - nu administrați doza necesară de doxorubicină dacă simptomele au apărut înainte de a 4-a zi a ciclului de tratament; pentru simptome după ziua 4, doza trebuie redusă cu 25% în ciclurile următoare dacă doza de bortezomib este redusă din cauza toxicității hematologice; nu administrați doza corectă de bortezomib. Dacă 2 sau mai multe doze de bortezomib sunt reținute într-un ciclu de tratament pentru ciclurile ulterioare, reduceți doza cu 25%. Dezvoltarea toxicității non-hematologice de gradul 3 sau 4 - nu administrați o doză de doxorubicină până când starea nu se îmbunătățește la copii și adolescenți de grad. Experiența la copii este limitată. Din acest motiv, nu se recomandă utilizarea la pacienții cu vârsta sub 18 ani. Grupuri speciale de pacienți. Pacienți cu insuficiență hepatică. Inițierea tratamentului - Dacă nivelul de bilirubină este cuprins între 1,2 - 3,0 mg / dL, prima doză trebuie redusă cu 25%. Dacă bilirubina este> 3,0 mg / dL, prima doză trebuie redusă cu 50%. Dacă pacientul tolerează prima doză fără creșterea bilirubinei sau a enzimelor hepatice, doza din cel de-al doilea ciclu poate fi crescută la nivelul dozei următoare, adică dacă prima doză este redusă cu 25%, atunci doza trebuie mărită la doza completă în al doilea ciclu; dacă prima doză este redusă cu 50%, doza trebuie crescută la 75% din valoarea sa completă în cel de-al doilea ciclu. Doza poate fi crescută la valoarea sa maximă în ciclurile următoare. La pacienții cu metastaze hepatice însoțite de o creștere a bilirubinei și a enzimelor hepatice, medicamentul poate fi utilizat de până la 4 ori limita superioară a normalului. Nu este necesară ajustarea dozelor la pacienții vârstnici și la pacienții cu insuficiență renală; nu sunt disponibile date farmacocinetice la pacienții cu CCr. Mod de administrare. Nu trebuie administrat pe cale intramusculară sau subcutanată. A nu se administra sub formă de bolus sau soluție nediluată. Se recomandă ca setul de perfuzie să fie conectat printr-o ramură laterală a cateterului la perfuzia intravenoasă de soluție de glucoză 5% (50 mg / ml) pentru a obține o diluție suplimentară și a minimiza riscul de tromboză și extravazare. Perfuzia poate fi administrată într-o venă periferică. Nu utilizați filtre de perfuzie în linie. Pentru doze <90 mg: diluați preparatul în 250 ml soluție perfuzabilă de glucoză 5% (50 mg / ml). Pentru doze ≥90 mg: diluați preparatul în 500 ml soluție perfuzabilă de glucoză 5% (50 mg / ml). Pentru cancerul de sân / cancer ovarian / mielom multiplu, prima doză trebuie administrată cu o rată de cel mult 1 mg / minut pentru a minimiza riscul de reacții la perfuzie. Dacă nu există nicio reacție legată de perfuzie, se pot administra perfuzii suplimentare în decurs de 60 de minute. Pacienții care prezintă o reacție la perfuzie trebuie modulați după cum urmează: 5% din doza totală trebuie perfuzată lent în primele 15 minute. Dacă perfuzia este tolerată fără răspuns, viteza de administrare poate fi dublată în următoarele 15 minute. Dacă perfuzia este încă tolerată, perfuzia poate fi oprită în decurs de o oră pentru un timp total de perfuzie de 90 de minute. În cazul sarcomului Kaposi în cursul SIDA, doza preparatului se diluează în 250 ml soluție de perfuzie de glucoză 5% (50 mg / ml) și se administrează ca perfuzie intravenoasă timp de 30 de minute.
Indicații
Monoterapie pentru cancerul de sân metastatic la pacienții cu risc crescut de complicații cardiace. Tratamentul cancerului ovarian avansat la pacienții a căror chimioterapie de primă linie cu compuși de platină a eșuat. Tratamentul pacienților cu progresie a mielomului multiplu în terapia combinată cu bortezomib care au primit cel puțin o linie de tratament anterioară și care au suferit deja sau nu sunt eligibili pentru transplant de măduvă osoasă. Tratamentul sarcomului Kaposi (KS) legat de SIDA la pacienții cu număr scăzut de CD4 (mai puțin de 200 / mm3) cu implicare semnificativă a membranelor mucoase, a pielii sau a organelor interne. Preparatul poate fi utilizat la pacienții cu SIDA-KS în chimioterapia de primă linie sau a doua linie, când a fost observată progresia bolii, în ciuda terapiei combinate utilizate anterior, care constă din cel puțin două dintre următoarele medicamente: alcaloizi vinca, bleomicină și forma farmaceutică standard de doxorubicină (sau altă antraciclină) sau fără toleranță.
Contraindicații
Hipersensibilitate la substanța activă, arahide sau soia. Nu trebuie utilizat la pacienții cu sarcom de SIDA Kaposi pentru care tratamentul local sau sistemic cu interferon alfa poate fi eficient.
Precauții
Datorită diferențelor dintre profilurile farmacocinetice și programele de dozare, preparatul nu trebuie utilizat în mod interschimbabil cu alte medicamente care conțin clorhidrat de doxorubicină. În timpul administrării medicamentului se recomandă teste ECG de rutină frecvente. Biopsia miocardică trebuie luată în considerare dacă apare o reducere a complexului QRS. În mod obișnuit, înainte de începerea tratamentului cu preparatul și repetat periodic în timpul tratamentului, se recomandă măsurarea ecocardiografică a fracției de ejecție a ventriculului stâng sau a angiografiei multi-cadru (MUGA). Evaluarea funcției ventriculare stângi este considerată obligatorie înainte de fiecare administrare suplimentară de medicament care depășește o doză cumulativă de 450 mg antracicline / m2. în timpul vieții. În timpul tratamentului cu antraciclină, cele menționate mai sustestele și metodele de evaluare a performanței cardiace trebuie utilizate în următoarea ordine: înregistrarea ECG, măsurarea fracției de ejecție a ventriculului stâng, biopsia endomiocardică. Datorită efectului cardiotoxic al medicamentului, trebuie acordată o atenție deosebită pacienților cu boli de inimă, inclusiv insuficiență cardiacă, și la pacienții care primesc alte antracicline. Doza totală de doxorubicină HCI trebuie să ia în considerare orice tratament anterior (sau concomitent) cu agenți cardiotoxici (inclusiv: alte antracicline, antrachinone sau de exemplu 5-fluorouracil); un grup de risc suplimentar sunt pacienții care au primit anterior iradiere mediastinală sau care primesc concomitent ciclofosfamidă, a căror cardiotoxicitate poate apărea și după o doză cumulativă de antracicline mai mică de 450 mg / m2. Profilul de siguranță cardiacă al regimului de doză recomandat pentru tratamentul cancerului de sân și ovarian (50 mg / m2) este similar cu cel al dozei de 20 mg / m2. la pacienții cu sarcom de Kaposi legat de SIDA. Datorită posibilității de tulburări ale măduvei osoase, ar trebui efectuate frecvente hemograme în timpul tratamentului (înainte de fiecare doză). Disfuncția persistentă a măduvei osoase poate duce la suprainfecții și sângerări. Au fost observate leucemii mieloide acute secundare și mielodisplazii la pacienții care au primit terapie combinată cu doxorubicină; orice pacient care primește doxorubicină trebuie să fie sub control hematologic. Datorită cazurilor de cancer oral secundar atât în timpul tratamentului, cât și până la 6 ani de la ultima doză, pacienții trebuie monitorizați în mod regulat pentru ulcerele gurii sau orice disconfort la nivelul gurii. Datorită posibilității unor reacții alergice și anafilactoide grave și uneori care pun viața în pericol la scurt timp după începerea perfuziei (cu simptome precum: astm, înroșire a urticariei, dureri în piept, febră, hipertensiune arterială, tahicardie, mâncărime, transpirație, dificultăți de respirație, umflături) frisoane, dureri de spate, strângere la nivelul pieptului și gâtului și / sau hipotensiune arterială, convulsii), prima doză trebuie administrată cu o rată de cel mult 1 mg / min. Fiecare flacon al preparatului conține zaharoză, iar medicamentul este administrat într-o soluție de glucoză 5%, care ar trebui luată în considerare la pacienții cu diabet zaharat. Acest medicament conține mai puțin de 1 mmol sodiu (23 mg) pe doză și este în esență „fără sodiu”.
Activitate nedorită
Cele mai frecvente efecte secundare observate în cancerul de sân sau ovarian sunt eritrodisestezia palmar-plantară - EPP (cazurile totale au fost de 44-46,1%; tratamentul a fost întrerupt la unii pacienți pentru EPP sever) și stomatita sau mucozita și greața . La pacienții cu sarcom de Kaposi legat de SIDA, disfuncția măduvei osoase (în principal leucopenie) a fost observată cel mai frecvent. La pacienții cu mielom multiplu, cele mai frecvent raportate reacții adverse (legate de tratament) în terapia combinată cu bortezomib au fost greață, diaree, neutropenie, trombocitopenie, vărsături, oboseală și constipație. Pacienți cu cancer de sân (doza preparatului 50 mg / m2 la fiecare 4 săptămâni). Foarte frecvente: anorexie, greață, stomatită, vărsături, EIP, alopecie, erupții cutanate, astenie, oboseală, mucozită nespecificată. Frecvente: faringită, leucopenie, anemie, neutropenie, trombocitopenie, parestezie, dureri abdominale, constipație, diaree, dispepsie, ulcerații ale gurii, piele uscată, decolorare a pielii, modificări ale pigmentării, eritem, erupție cutanată, slăbiciune, pierexie, durere, foliculită , infecții fungice, herpes al buzelor (origine non-herpetică), infecții ale căilor respiratorii superioare, neuropatie periferică, lacrimare, vedere încețoșată, aritmie ventriculară, epistaxis, durere orală, erupții buloase, dermatită, erupții cutanate eritematoase, boli ale unghiilor, solzoase piele, crampe la nivelul picioarelor, dureri osoase, dureri musculo-scheletice, dureri ale sânilor, edeme, umflături ale picioarelor. Mai puțin frecvente: somnolență. Pacienți cu cancer ovarian (doza preparatului 50 mg / m2 la fiecare 4 săptămâni). Foarte frecvente: leucopenie, anemie, neutropenie, trombocitopenie, anorexie, constipație, diaree, greață, stomatită, vărsături, eritrodisestezie palmar-plantară (sindromul mână-picior; PPE), alopecie, erupție cutanată, slăbiciune, tulburări ale mucoasei. Frecvente: faringită, parestezie, somnolență, durere abdominală, dispepsie, ulcerații bucale, piele uscată, decolorare a pielii, pierexie, durere, infecție, candidoză orală, zona zoster, infecție a tractului urinar, anemie hipocromă, reacții alergice, deshidratare, cașexie, anxietate , depresie, insomnie, cefalee, amețeli, neuropatie, hipertensiune arterială, conjunctivită, tulburări cardiovasculare, vasodilatație, dispnee, creșterea tusei, ulcerații bucale, esofagită, gastrită, disfagie, gură uscată, flatulență, gingivită, disgeuzie, erupție veziculară, mâncărime, dermatită exfoliativă, modificări ale pielii, erupție maculopapulară, transpirație, acnee, ulcerații ale pielii, dureri de spate, dureri musculare, urinare dureroasă, vaginite, frisoane, dureri toracice, stare de rău, edem periferic, scădere în greutate. Pacienți cu mielom multiplu (doză de 30 mg / m2 de preparat în asociere cu bortezomib într-un ciclu de 3 săptămâni). Foarte frecvente: anemie, neutropenie, trombocitopenie, anorexie, neuropatie senzorială periferică, nevralgie, cefalee, greață, diaree, vărsături, constipație, stomatită, EPP, erupție cutanată, astenie, oboseală, febră. Frecvente: herpes, herpes zoster, leucopenie, scăderea poftei de mâncare, insomnie, neuropatie periferică, neuropatie, parestezie, polineuropatie, amețeli, disgeuzie, dispnee, dureri abdominale, dispepsie, piele uscată, durere la nivelul extremităților, scădere în greutate, pneumonie, nazofaringită , infecții ale căilor respiratorii superioare, candidoză orală, neutropenie febrilă, limfopenie, deshidratare, hipokaliemie, hiperkaliemie, hipomagnezemie, hiponatremie, hipocalcemie, anxietate, letargie, hipoestezie, sincopă, tulburări senzoriale, conjunctivită, hipotensiune arterială, hipotensiune arterială, hipotensiune arterială roșeață a pielii, hipertensiune arterială, flebită, tuse, epistaxis, dispnee la efort, durere în tractul gastro-intestinal superior, ulcerație a gurii, gură uscată, disfagie, stomatită aftoasă, prurit, urticarie papulară, dermatită alergică, eritem, hiperpigmentare a pielii, echimoze asemănătoare punctelor, alopecie, erupție pe piele, artralgie, mialgie, spasme musculare, slăbiciune musculară, dureri musculo-scheletice, dureri musculo-scheletice în piept, eritem scrotal, edem periferic, frisoane, simptome para-gripale, stare de rău, hipertermie, niveluri crescute de AST , scăderea fracției de ejecție miocardică, creșterea creatininei, creșterea ALT. Pacienți cu sarcom Kaposi în cursul SIDA (doza de preparare 20 mg / m2 la 2-3 săptămâni). Foarte frecvente: neutropenie, anemie, leucopenie, greață. Frecvente: candidoză orală, trombocitopenie, anorexie, amețeli, retinită, vasodilatație, dispnee, diaree, gastrită, vărsături, ulcerații bucale, dureri abdominale, glossită, constipație, greață, vărsături, alopecie, erupție cutanată, slăbiciune, febră, reacții acute la perfuzie, scădere în greutate. Mai puțin frecvente: confuzie, tulburări senzoriale, eritem al palmelor și tălpilor (EPP). Au fost observate și reacții de hipersensibilitate, inclusiv reacții anafilactice (infecții cu Pneumocystis carinii, infecții cu Mycobacterium avium) și frecvent observate la pacienții cu imunodeficiență indusă de HIV. Toate grupurile de pacienți. Reacții legate de perfuzie: reacții de hipersensibilitate, reacții anafilactoide, bronhospasm, umflături faciale, hipotensiune arterială, vasodilatație, urticarie, dureri de spate, dureri toracice, frisoane, febră, hipertensiune arterială, tahicardie, dispepsie, greață, amețeli, dificultăți respiratorii, faringită, erupție cutanată, mâncărime, transpirație, reacții la locul injectării și interacțiuni medicamentoase. Foarte rar, au fost raportate convulsii în asociere cu reacții legate de perfuzie. Toți pacienții au prezentat reacții legate de perfuzie, în principal în timpul primei perfuzii. Oprirea temporară a perfuziei corectează de obicei aceste simptome fără a necesita tratament suplimentar. La aproape toți pacienții, tratamentul cu preparatul poate fi reluat după ce simptomele au dispărut fără să se repete. Reacțiile legate de perfuzie apar rar cu ciclurile de tratament ulterioare. A fost raportată disfuncția măduvei osoase care duce la anemie, trombocitopenie, leucopenie și, rareori, neutropenie febrilă. Stomatita a fost raportată frecvent la pacienții cărora li se administrează perfuzie continuă. O creștere a incidenței CHF a fost observată cu tratamentul cu doxorubicină la o doză cumulativă> 450 mg / m2. în viață sau la o doză mai mică la pacienții cu risc de a dezvolta complicații ale mușchiului cardiac. Au fost observate leucemii mieloide acute secundare și mielodisplazie la pacienții care au primit terapie combinată cu doxorubicină. Modificări necrotice locale care rezultă din extravazare au fost observate foarte rar (în caz de simptome, perfuzia trebuie oprită imediat și restul medicamentului administrat într-o altă venă). Rar, a apărut reapariția leziunilor cutanate din cauza radioterapiei anterioare. Boli severe ale pielii (eritemul multiform, sindromul Stevens-Johnson și necroliza epidermică toxică) au fost raportate foarte rar în experiența după punerea pe piață. Au fost raportate cazuri rare de tromboembolism venos, inclusiv tromboflebită, tromboză venoasă și embolie pulmonară (deoarece pacienții cu cancer prezintă un risc mai mare de tromboembolism, nu se poate stabili o relație de cauzalitate cu utilizarea preparatului). Modificările necrotice rezultate din extravazare au fost foarte rar observate.
Sarcina și alăptarea
Nu utilizați în timpul sarcinii, cu excepția cazului în care este absolut necesar (există riscul apariției unor malformații congenitale severe la făt). Femeile aflate la vârsta fertilă trebuie să evite să rămână gravide în timp ce ele sau partenerii lor sunt tratați și timp de 6 luni după oprirea tratamentului. Alăptarea trebuie întreruptă înainte de începerea tratamentului cu preparatul. Femeile infectate cu HIV nu trebuie să-și alăpteze bebelușii în niciun caz pentru a evita transmiterea de la mamă la copil.
Comentarii
Preparatul nu are nicio influență sau are o influență neglijabilă asupra capacității de a conduce vehicule și de a folosi utilaje. Cu toate acestea, pacienții trebuie să evite să conducă vehicule sau să folosească utilaje dacă prezintă amețeli sau somnolență.
Interacțiuni
Se recomandă prudență la administrarea concomitentă a medicamentelor care interacționează cu clorhidratul standard de doxorubicină. Preparatul poate crește toxicitatea altor tratamente anti-cancer. Nu a fost observată nicio toxicitate suplimentară la pacienții cu tumori solide (inclusiv cancer de sân și ovarian) tratați concomitent cu ciclofosfamidă sau taxani în timpul studiilor clinice. La pacienții cu SIDA, s-a raportat că clorhidratul de doxorubicină standard potențează cistita hemoragică indusă de ciclofosfamidă și potențează hepatotoxicitatea 6-mercaptopurinei. Se recomandă prudență la utilizarea oricărui alt medicament citotoxic în același timp, în special a oricărui medicament care dăunează funcției măduvei osoase.
Preparatul conține substanța: clorhidrat de doxorubicină
Medicament rambursat: NU





-przyczyny-objawy-leczenie.jpg)


-gowy---badanie-rtg-mzgu.jpg)





---norma-wyniki.jpg)

---zastosowanie-i-dawkowanie.jpg)