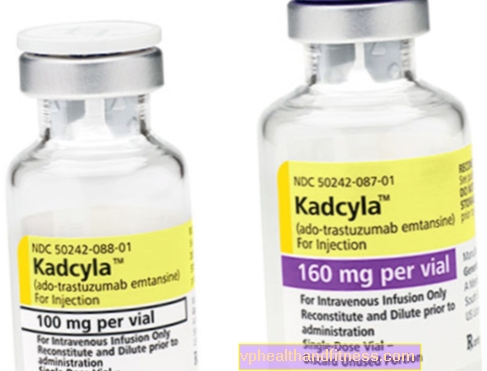
1 flacon conține 100 mg (160 mg) pulbere pentru reconstituire 5 ml (8 ml) concentrat de trastuzumab emtansină la o concentrație de 20 mg / ml pentru perfuzie după reconstituire.
| Nume | Conținutul pachetului | Substanța activă | Preț 100% | Modificat ultima dată |
| Kadcyla | 1 flacon, pulbere pentru preparare Soluție finală a inf. | Trastuzumab emtansine | 2019-04-05 |
Acțiune
Medicament anticancerigen. Trastuzumab emtansina este un conjugat anticorp-medicament care conține trastuzumab, un anticorp monoclonal anti-HER2 IgG1 umanizat, legat covalent de DM1 (derivat de maytansină, inhibitor de microtubuli) printr-o legătură tioeter MCC stabilă (4- ciclohexan-1-carbox . Emtansine este complexul MCC-DM1. Conjugarea DM1 cu trastuzumab determină acțiunea selectivă a medicamentelor citotoxice împotriva celulelor tumorale supraexprimate HER2, crescând astfel concentrația intracelulară a DM1 direct în celulele tumorale. După legarea la HER2, trastuzumab emtansina este internalizarea mediată de receptor urmată de degradarea lizozomală, eliberând cataboliți care conțin DM1 (în principal lizină-MCC-DM1). Mecanismul de acțiune al trastuzumab emtansine este mediat de activitatea trastuzumab și DM1. Trastuzumab emtansina, ca și trastuzumab, se leagă de domeniul IV al domeniului extracelular (ECD) al receptorului, precum și de receptorii Fcγ și complementul C1q. Mai mult, inhibă activitatea domeniului ECD al receptorului HER2, inhibă semnalizarea căii fosfatidilinozitol 3-kinazei (PI3-K) și mediază citotoxicitatea celulară dependentă de anticorpi (ADCC) în celulele cancerului de sân uman care supraexprimă HER2. DM1 se leagă de tubulină. Prin inhibarea polimerizării tubulinei, atât DM1 cât și trastuzumab emtansina determină arestarea celulelor în faza G2 / M a ciclului celular, ducând în cele din urmă la moartea celulelor prin apoptoză. Linkerul MCC reduce eliberarea sistemică de DM1 și crește concentrația sa la locul țintă. Trastuzumab emtansina este deconjugată și apoi catabolizată prin proteoliză în lizozomii celulari. DM1 este metabolizat în principal de CYP3A4 și într-o măsură mai mică de CYP3A5. T0.5 al trastuzumab este de aproximativ 4 zile. Nu a existat nicio acumulare de trastuzumab emtansină după mai multe perfuzii intravenoase administrate la un interval de 3 săptămâni. Vârsta nu a avut niciun efect asupra farmacocineticii trastuzumab emtansinei.
Dozare
Intravenos. Preparatul trebuie prescris de un medic și administrat sub supravegherea cadrelor medicale cu experiență în tratarea pacienților cu cancer. Pacienții cărora li s-a administrat trastuzumab emtansină ar trebui să aibă cancer HER2 pozitiv - scor imunohistochimie (IHC) 3+ sau raport ≥2 la hibridizarea in situ (ISH). Testele trebuie efectuate folosind teste de diagnostic in vitro (IVD) cu marcaj CE. În cazul în care testul CE IVD nu este disponibil, testul trebuie efectuat cu un alt test validat. Pentru a evita erorile medicale, este important să verificați eticheta flaconului pentru a vă asigura că medicamentul preparat și administrat este Kadcyla (trastuzumab emtansină) și nu Herceptin (trastuzumab). Doza recomandată este de 3,6 mg / kg. administrat sub formă de perfuzie intravenoasă la fiecare 3 săptămâni (ciclul de 21 de zile). Pacienții trebuie tratați până la progresia tumorii sau până la obținerea unei toxicități inacceptabile. Doza inițială trebuie administrată sub formă de perfuzie intravenoasă de 90 de minute. Locul injectării trebuie monitorizat îndeaproape, deoarece medicamentul poate pătrunde subcutanat în timpul administrării. Dacă perfuzia anterioară a fost bine tolerată, dozele ulterioare pot fi administrate sub formă de perfuzie de 30 de minute. Pacienții trebuie observați în timpul perfuziei și timp de cel puțin 30 de minute după aceea. Viteza perfuziei trebuie încetinită sau întreruptă dacă pacientul dezvoltă simptome legate de perfuzie. Trastuzumab emtansină trebuie întrerupt în cazul reacțiilor la perfuzie care pun viața în pericol. Medicamentele pentru reacțiile alergice / anafilactice la perfuzie, precum și echipamentele de urgență trebuie să fie disponibile pentru utilizare imediată. Dacă se omite o doză planificată, aceasta trebuie administrată cât mai curând posibil; nu așteptați până la următorul ciclu. Programul de dozare trebuie ajustat pentru a menține intervalul de dozare de 3 săptămâni. Următoarea doză trebuie administrată conform recomandărilor de dozare. Tratamentul reacțiilor adverse simptomatice poate include întreruperea periodică, reducerea dozei sau întreruperea tratamentului. Schema de reducere a dozei (doza inițială 3,6 mg / kg): prima reducere a dozei 3 mg / kg; a doua reducere a dozei 2,4 mg / kg corp; dacă este necesară reducerea suplimentară a dozei, tratamentul trebuie oprit. Liniile directoare privind modificarea dozelor pentru creșteri AST și ALT: Gradul 2 (> 2,5 până la ≤5 x ULN) - nu este necesară ajustarea dozei; gradul 3 (> 5 până la ≤20 x ULN) - utilizați preparatul dacă AST / ALT scade la gradul ≤ 2 (> 2,5 până la 20 x ULN) - întrerupeți tratamentul. Reguli de modificare a dozei pentru hiperbilirubinemie: gradul 2 (> 1,5 până la ≤3 x ULN) - utilizați preparatul atunci când nivelul de bilirubină a scăzut la gradul ≤1. (> ULN la 1,5 x ULN), nu este necesară ajustarea dozei; Gradul 3 (> 3 până la ≤10 x ULN) - utilizați preparatul dacă concentrația de bilirubină scade la gradul ≤ 1 (> ULN la 1,5 x ULN), apoi reduceți doza (vezi schema de reducere a dozei); Gradul 4 (> 10 x ULN) - Tratamentul final. Reguli de modificare a dozei pentru trombocitopenie: gradul 3 (concentrația trombocitelor 25.000 până la 3) - utilizați preparatul atunci când concentrația trombocitelor este ≤1. (de exemplu, ≥75.000 / mm3); nu este necesară ajustarea dozei; Gradul 4 (concentrația de trombocite 3) - utilizați preparatul atunci când concentrația de trombocite atinge ≤1. (de ex. ≥75.000 / mm3) apoi reduceți doza (vezi schema de reducere a dozei). Principii de modificare a dozelor la pacienții cu disfuncție ventriculară: LVEF 45% - continuați terapia; LVEF 40% până la ≤45% cu o reducere simultană a fracției de ejecție cu - nu utilizați preparatul; Reevaluează FEVS în termen de 3 săptămâni, dacă FEVS este încă ≥ 10 puncte procentuale față de momentul inițial, întrerupeți tratamentul; CHF simptomatică - opriți tratamentul. Copii și adolescenți: Siguranța și eficacitatea la copii și adolescenți cu vârsta sub 18 ani nu au fost stabilite, deoarece cancerul de sân diseminat (MBC) nu este cunoscut la această populație. Grupuri speciale de pacienți. Terapia trebuie întreruptă temporar la pacienții care dezvoltă neuropatie periferică de gradul 3 sau 4 până când atinge gradul ≤2; la reluarea tratamentului, se poate lua în considerare reducerea dozei așa cum este prezentat în schema de reducere a dozei. Nu este necesară ajustarea dozei la pacienții cu vârsta ≥ 65 de ani; nu există date suficiente pentru a determina siguranța și eficacitatea terapiei la pacienții cu vârsta ≥75 de ani. Nu este necesară ajustarea dozei inițiale la pacienții cu insuficiență renală ușoară sau moderată; Necesitatea potențială de ajustare a dozei la pacienții cu insuficiență renală severă nu poate fi determinată și, prin urmare, acești pacienți trebuie monitorizați cu atenție. Nu este necesară ajustarea dozei inițiale la pacienții cu insuficiență hepatică ușoară sau moderată. Trastuzumab emtansină nu a fost studiat la pacienții cu insuficiență hepatică severă; trebuie administrată precauție la tratarea pacienților cu insuficiență hepatică din cauza hepatotoxicității cunoscute observate în timpul tratamentului. Mod de a da. Preparatul trebuie dizolvat și diluat de personalul medical și administrat sub formă de perfuzie intravenoasă. Nu trebuie administrat sub formă de bolus sau injecție rapidă.
Indicații
Monoterapie a pacienților adulți cu cancer de sân HER2-pozitiv, inoperabil local sau metastatic, tratat anterior cu trastuzumab și un taxan, în combinație sau separat. Pacienții care au urmat anterior tratament pentru boală locală avansată sau generalizată sau care au recidivat în timpul sau în termen de 6 luni de la finalizarea terapiei adjuvante.
Contraindicații
Hipersensibilitate la substanța activă sau la alte ingrediente ale preparatului.
Precauții
În studiile clinice cu trastuzumab emtansină au fost raportate cazuri de boli pulmonare interstițiale (ILD), inclusiv pneumonie; unele au fost asociate cu insuficiență respiratorie acută sau au fost fatale. Întreruperea tratamentului cu preparatul este recomandată la pacienții diagnosticați cu ILD sau pneumonie. Pacienții cu dispnee în repaus, asociați cu complicații ale cancerului avansat și boli asociate pot prezenta un risc crescut de a dezvolta complicații pulmonare. În timpul tratamentului cu preparatul, s-au observat hepatotoxicitate și tulburări severe ale ficatului și ale tractului biliar, inclusiv hipertrofie regenerativă nodulară (NRH) a ficatului și decese datorate leziunilor hepatice induse de medicamente; comorbiditățile și / sau medicamentele concomitente cunoscute ca având potențial hepatotoxic pot fi, de asemenea, afectate. Funcția hepatică trebuie monitorizată înainte de inițierea tratamentului și înainte de administrarea ulterioară. Pacienții cu o creștere inițială a ALT (de exemplu, asociați cu metastaze hepatice) sunt predispuși la dezvoltarea insuficienței hepatice și prezintă un risc mai mare de toxicitate hepatică de grad 3-5 sau niveluri crescute de laborator hepatice. Cazurile de NRH au fost diagnosticate prin efectuarea de biopsii hepatice. Apariția NRH trebuie luată în considerare la toți pacienții cu semne clinice de hipertensiune portală și / sau imagine asemănătoare cirozei observate la CT hepatică, dar cu niveluri serice normale de transaminază și fără alte dovezi ale cirozei. Dacă este diagnosticat NRH, tratamentul cu preparatul trebuie întrerupt. Nu a fost studiat la pacienți cu transaminaze serice> 2,5 x LSN (limita superioară a normalului) sau cu bilirubină totală> 1,5 x LSN înainte de inițierea tratamentului. În cazul activității transaminazelor serice> 3 x ULN și simultan concentrația totală de bilirubină> 2 x ULN, tratamentul trebuie întrerupt. Se recomandă prudență în tratarea pacienților cu insuficiență hepatică. Există un risc crescut de disfuncție ventriculară stângă în timpul tratamentului. La pacienții tratați cu trastuzumab emtansină, a fost observată o reducere a fracției de ejecție a ventriculului stâng (FEVS) la 50 de ani), cu o valoare inițială scăzută a FEVS (25 kg / m2). Testele standard ale funcției cardiace (ecocardiogramă sau angiografie cu radionuclizi multi-gate) trebuie efectuate înainte de inițierea tratamentului și la intervale regulate (de exemplu, la fiecare 3 luni). FEVS la pacienți la momentul inițial a fost ≥50% în majoritatea studiilor clinice. Pacienții cu insuficiență cardiacă congestivă, aritmii severe care necesită tratament, antecedente de infarct miocardic sau angină instabilă în decurs de 6 luni înainte de randomizare sau cu dispnee în repaus din cauza cancerului avansat, au fost excluși din studiu. În cazul tulburărilor ventriculare stângi, administrarea următoarei doze trebuie amânată sau tratamentul trebuie întrerupt. Efectul tratamentului cu trastuzumab emtansină la pacienții care întrerup tratamentul cu trastuzumab din cauza unei reacții legate de perfuzie nu a fost studiat; terapia cu acest medicament nu este recomandată la acest grup de pacienți. Terapia trebuie întreruptă la pacienții care dezvoltă o reacție severă legată de perfuzie până când simptomele se remit. Reinițierea terapiei trebuie luată în considerare pe baza judecății clinice a severității reacției. Tratamentul trebuie întrerupt în cazul unei reacții la perfuzie care pune viața în pericol. Efectul tratamentului cu trastuzumab emtansină la pacienții care au întrerupt tratamentul cu trastuzumab din cauza hipersensibilității nu a fost studiat; Nu se recomandă utilizarea trastuzumab emtansinei la acești pacienți. Pacienții trebuie monitorizați pentru hipersensibilitate / reacții alergice, simptomele pot fi aceleași cu reacțiile legate de perfuzie; au fost observate reacții anafilactice severe. Dacă apare hipersensibilitate (cu reacții crescute în timpul perfuziilor ulterioare), tratamentul cu trastuzumab emtansină trebuie întrerupt.Datorită riscului de evenimente hemoragice (incluzând hemoragia SNC, respiratorie și gastro-intestinală), este necesară prudență și trebuie luată în considerare monitorizarea suplimentară atunci când se administrează concomitent cu anticoagulante sau medicamente antiagregante. Se recomandă monitorizarea numărului de trombocite înainte de fiecare doză de trastuzumab emtansină. Pacienții cu trombocitopenie (≤100.000 / mm3) și pacienții care primesc terapie anticoagulantă (de exemplu, warfarină, heparină, heparine cu greutate moleculară mică) trebuie monitorizați îndeaproape în timpul tratamentului cu trastuzumab emtansină. Trastuzumab emtansină nu a fost studiat la pacienții cu număr de trombocite ≤100.000 / mm3 înainte de inițierea tratamentului. În cazul înrăutățirii trombocitopeniei la gradul 3 sau mai mare (3), preparatul nu trebuie utilizat până când toxicitatea scade la gradul 1 (≥75.000 / mm3). În studiile clinice cu trastuzumab emtansină, a apărut neuropatia periferică de gradul 1, în principal senzorială. Pacienții cu neuropatie periferică de grad ≥3 au fost excluși de la participarea la studiile clinice. Tratamentul pacienților cu neuropatie periferică de gradul 3 sau 4 trebuie întrerupt periodic până când complicația este redusă la gradul ≤2. Pacienții trebuie monitorizați în mod regulat pentru a observa semne de neurotoxicitate. Pentru a îmbunătăți trasabilitatea preparatelor biologice, denumirea comercială a medicamentului administrat trebuie să fie scrisă clar (sau menționată) în dosarul pacientului. Siguranța și eficacitatea medicamentului la copii și adolescenți cu vârsta sub 18 ani nu au fost stabilite, deoarece cancerul de sân diseminat nu se găsește la această populație. Nu trebuie administrat sub formă de bolus sau injecție rapidă.
Activitate nedorită
Foarte frecvente: infecții ale tractului urinar, trombocitopenie, anemie, hipokaliemie, insomnie, neuropatie periferică, cefalee, sângerări, epistaxis, tuse, dispnee, stomatită, diaree, vărsături, greață, constipație, gură uscată, dureri abdominale , erupții cutanate, dureri musculo-scheletice, artralgii, mialgii, oboseală, febră, astenie, frisoane, creșterea transaminazelor. Frecvente: neutropenie, leucopenie, hipersensibilitate, amețeli, disgeuzie, tulburări de memorie, sindrom de ochi uscat, conjunctivită, tulburări vizuale, creșterea lacrimării, disfuncție ventriculară stângă, hipertensiune arterială, dispepsie, sângerare gingivală, prurit cutanat, alopecie, boală a unghiilor , eritrodisestezie palmar-plantară, urticarie, edem periferic, creșterea fosfatazei alcaline, reacții legate de perfuzie (roșeață a pielii, frisoane, febră, dispnee, tensiune arterială scăzută, respirație șuierătoare, bronhospasm, tahicardie). Mai puțin frecvente: pneumonie (ILD), hepatotoxicitate, insuficiență hepatică, hiperplazie nodulară regenerativă, hipertensiune portală, extravazare la locul injectării (eritem, sensibilitate, iritație a pielii, durere sau umflături). În plus, a fost observată hiperbilirubinemie. În studiile clinice, creșteri ale transaminazelor serice (gradul 1-4) au apărut în timpul tratamentului cu trastuzumab emtansină și au fost, în general, tranzitorii. Creșterea transaminazelor a fost cel mai adesea tranzitorie, atingând vârful în a 8-a zi după administrare. Ulterior, toxicitatea a scăzut la gradul 1 sau s-a rezolvat înainte de următorul ciclu. A existat, de asemenea, un efect cumulativ (procentul pacienților cu creștere ALT / AST de gradul 1-2 a crescut odată cu ciclurile ulterioare). La pacienții cu transaminaze crescute, majoritatea pacienților au prezentat reducere sau recuperare a toxicității de gradul 1 în decurs de 30 de zile de la ultima doză de trastuzumab emtansină. Disfuncția ventriculară stângă a fost găsită la 2,2% dintre pacienții care au participat la studii clinice. În majoritatea cazurilor, reducerile FEVS de gradul 1 sau 2 au fost asimptomatice.Toxicitatea de gradul 3 sau 4 a fost observată la 0,4% dintre pacienți, de obicei în timpul ciclurilor inițiale de tratament (1-2). Au fost raportate evenimente hemoragice severe (grad ≥3) la 2,2% dintre toți pacienții. Trombocitopenia a apărut la 24,9% dintre pacienți în studiile clinice și a fost cea mai frecventă reacție adversă care a dus la întreruperea tratamentului. În studiile clinice, incidența și severitatea trombocitopeniei au fost mai mari la pacienții asiatici. Unii pacienți care au dezvoltat aceste complicații au fost tratați și cu terapie anticoagulantă. Au existat episoade fatale de sângerare și complicații severe de sângerare, inclusiv sângerări ale SNC 5,3% dintre pacienți au testat pozitiv pentru anticorpi anti-trastuzumab emtansină.
Sarcina și alăptarea
Nu este recomandată utilizarea trastuzumab emtansinei la femeile gravide. Înainte de sarcină, femeile ar trebui să fie informate cu privire la posibilitatea de a dăuna fătului. Cu toate acestea, pacientele care rămân însărcinate trebuie să contacteze imediat un medic. Dacă o femeie însărcinată este tratată cu trastuzumab emtansină, se recomandă monitorizarea atentă de către o echipă multidisciplinară. Trastuzumab poate provoca leziuni fetale sau moarte dacă este administrat unei femei gravide. La copiii gravidelor tratați cu preparatul, au fost raportate cazuri de oligohidramnios, unele dintre ele ducând la hipoplazie pulmonară fatală. DM1, componenta citotoxică a trastuzumab emtansinei, poate fi teratogenă și potențial embriotoxică. Femeile trebuie să oprească alăptarea înainte de a începe tratamentul cu trastuzumab emtansină. Pacienții pot începe să alăpteze la 7 luni după întreruperea tratamentului. Femeile aflate la vârsta fertilă trebuie să utilizeze contracepție eficientă în timpul tratamentului cu medicamentul și timp de 7 luni după ultima doză de trastuzumab emtansină. De asemenea, bărbații sau partenerele lor de sex feminin ar trebui să utilizeze metode contraceptive eficiente.
Comentarii
Pacienții care prezintă reacții legate de perfuzie nu trebuie să conducă vehicule sau să folosească utilaje până când aceste simptome nu vor fi rezolvate. Informații pregătite pe baza SPC de 13.07.2017 SmPC-ul actual este disponibil la www.roche.pl.
Interacțiuni
Rezultatele studiilor de metabolizare in vitro la microzomi hepatici umani indică faptul că DM1 este metabolizat în principal de enzima CYP3A4 și într-o măsură mai mică de CYP3A5. Utilizarea concomitentă a inhibitorilor puternici ai CYP3A4 (de exemplu, ketoconazol, itraconazol, claritromicină, atazanavir, indinavir, nefazodonă, nelfinavir, ritonavir, saquinavir, telitromicină și voriconazol) trebuie evitată, deoarece acest lucru poate crește nivelul DM1 și toxicitatea. Trebuie luată în considerare o formulare alternativă care nu inhibă sau doar inhibă ușor CYP3A4. Dacă utilizarea concomitentă a inhibitorilor puternici ai CYP3A4 nu poate fi evitată și, acolo unde este posibil, luați în considerare întârzierea administrării trastuzumab emtansinei până când inhibitorii CYP3A4 sunt eliminați din circulație (timp de înjumătățire aproximativ 3 inhibitori). Cu toate acestea, dacă se utilizează concomitent un inhibitor puternic al CYP3A4, iar tratamentul cu trastuzumab emtansină nu poate fi întârziat, pacientul trebuie monitorizat îndeaproape pentru efecte adverse în astfel de cazuri.
Preparatul conține substanța: Trastuzumab emtansină
Medicament rambursat: NU










---przyczyny-objawy-i-leczenie.jpg)