1 ml de soluție conține 100 mg de imunoglobulină normală umană (inclusiv cel puțin 95% IgG). Distribuția subclaselor IgG: IgG1 - aproximativ 60%, IgG2 - aproximativ 32%, IgG3 - aproximativ 7%, IgG4 - aproximativ 1%. Conținutul maxim de IgA nu este mai mare de 0,4 mg. PH-ul soluției este de 4,5 - 5,0; osmolalitatea sa este ≥ 240 mOsmol / kg. Acest medicament conține nu mai mult de 0,03 mmol (0,69 mg) sodiu per ml de soluție. Preparatul conține maltoză.
| Nume | Conținutul pachetului | Substanța activă | Preț 100% | Modificat ultima dată |
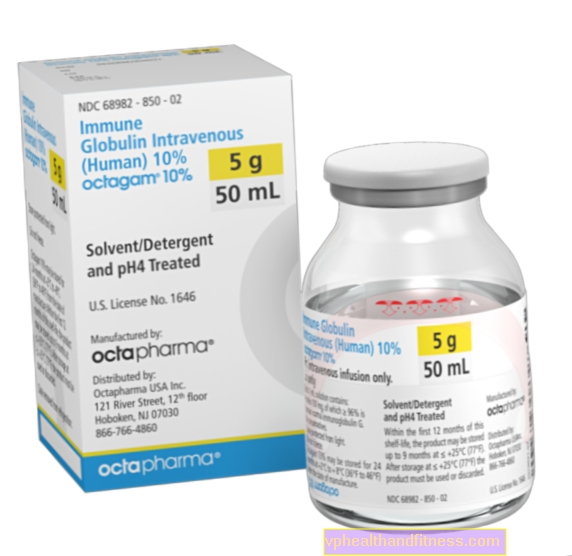
| Octagam 10% | pantof. 200 ml, sol. a inf. | Imunoglobulină umană normală | 2019-04-05 |
Acțiune
Imunoglobulina umană normală constă în principal din imunoglobulină G (IgG) neschimbată funcțional, cu un spectru larg de anticorpi împotriva agenților infecțioși. Are o distribuție a subclaselor de imunoglobulină G strâns proporționale cu cea din plasma umană nativă. Dozele suficient de mari de imunoglobulină umană normală pot readuce nivelurile anormal de scăzute de imunoglobulină G în intervalul normal. Mecanismul de acțiune al medicamentului administrat pe alte indicații decât terapia de substituție nu este complet elucidat, dar se știe că implică efecte imunomodulatoare. Imunoglobulina umană normală este imediat și complet biodisponibilă în circulația destinatarului după administrarea intravenoasă. Se distribuie relativ repede între plasmă și fluidul extracelular; după 3-5 zile se stabilește o stare de echilibru între compartimentele intra și extra-vasculare. Timpul de înjumătățire plasmatică pentru IgG la pacienții imunocompromiși după administrarea preparatului variază între 26 și 41 de zile. Timpul de înjumătățire poate varia de la pacient la pacient, în special în cazurile de imunodeficiență primară. Imunoglobulina G (IgG) și complexele sale sunt degradate în celulele sistemului reticuloendotelial.
Dozare
Intravenos. Adulți și copii (0-18 ani). Terapia de substituție trebuie inițiată și monitorizată sub supravegherea unui medic cu experiență în tratamentul imunodeficienței. Doza și regimul de dozare depind de indicație. În terapia de substituție, poate fi necesară individualizarea dozei pentru pacient, în funcție de răspunsul farmacocinetic și clinic. Următoarele scheme de dozare sunt orientări. Terapia de substituție în imunodeficiențele primare. Regimul de dozare trebuie să atingă niveluri minime de IgG (măsurate înainte de următoarea perfuzie) de cel puțin 5-6 g / l. Este nevoie de 3-6 luni pentru a corecta deficiența de anticorpi după începerea tratamentului. Doza inițială recomandată este de 0,4-0,8 g / kg. administrat o dată, apoi se utilizează 0,2 g / kg greutate corporală. la fiecare 3-4 săptămâni Este necesară o doză de 0,2-0,8 g / kg greutate corporală / lună pentru a atinge niveluri de IgG de 5-6 g / l. Intervalul dintre doze după atingerea unei stări de echilibru este de 3-4 săptămâni. Determinarea și evaluarea concentrațiilor minime trebuie făcute în raport cu incidența infecției. S-ar putea să fie necesar să se mărească doza și să se urmărească niveluri mai scăzute pentru a reduce rata infecției. Hipogammaglobulinemia și infecțiile bacteriene recurente la pacienții cu leucemie limfocitară cronică pentru care terapia profilactică cu antibiotice nu a reușit să producă rezultate satisfăcătoare; hipogammaglobulinemie și infecții bacteriene recurente la pacienții cu fază de platou cu mielom multiplu care nu au reușit să răspundă la imunizarea pneumococică; SIDA congenitală cu infecții bacteriene recurente: doza recomandată este de 0,2-0,4 g / kg. la 3-4 săptămâni Hipogammaglobulinemie la pacienți după transplant alogen de celule stem hematopoietice: doza recomandată este de 0,2-0,4 g / kg la 3-4 săptămâni. Nivelurile minime trebuie menținute peste 5 g / l. Trombocitopenie imună primară. Există două scheme de tratament alternative: 0,8-1 g / kg în ziua 1 a tratamentului; această doză poate fi repetată o dată în decurs de 3 zile sau 0,4 g / kg / 24 ore timp de 2-5 zile. Tratamentul poate fi repetat dacă boala reapare. Sindromul Guillain Barre: 0,4 g / kg greutate corporală zilnic timp de 5 zile. Boala Kawasaki: 1,6-2 g / kg în doze divizate timp de 2-5 zile sau o doză unică de 2 g / kg greutate corporală. Pacienții trebuie tratați cu acid acetilsalicilic. Mod de a da. Se administrează intravenos sub formă de perfuzie la o rată inițială de 0,01 ml / kg greutate corporală / min timp de 30 de minute. Dacă este bine tolerat, viteza de administrare poate fi crescută treptat până la maxim 0,12 ml / kg / min.
Indicații
Terapia de substituție la adulți, copii și adolescenți (0-18 ani). Sindroame de imunodeficiență primară cu producție afectată de anticorpi. Hipogammaglobulinemia și infecțiile bacteriene recurente la pacienții cu leucemie limfocitară cronică pentru care terapia profilactică cu antibiotice nu a reușit să producă rezultate satisfăcătoare. Hipogamaglobulinemie și infecții bacteriene recurente la pacienții cu mielom multiplu în fază de platou care nu au reușit să răspundă la imunizarea pneumococică. Hipogamaglobulinemie la pacienți după transplant alogen de celule stem hematopoietice (HSCT). SIDA congenital cu infecții bacteriene recurente. Imunomodulare la adulți și copii și adolescenți (0-18 ani). Trombocitopenia imună primară (ITP) la pacienții cu risc crescut de sângerare sau înainte de intervenția chirurgicală pentru creșterea numărului de trombocite în sânge. Echipa Guillain Barré. Boala Kawasaki.
Contraindicații
Hipersensibilitate la substanța activă sau la oricare dintre excipienți. Hipersensibilitate la imunoglobuline umane, în special la pacienții cu anticorpi împotriva IgA.
Precauții
Este esențial să urmați instrucțiunile de dozare și să respectați cu strictețe viteza de perfuzare prescrisă. Pacienții trebuie monitorizați cu atenție pentru efectele secundare în timpul perfuziei. Anumite reacții adverse pot fi mai frecvente la o rată de perfuzie ridicată sau la pacienții cărora li se administrează pentru prima dată imunoglobulină normală umană sau, în cazuri rare, când se trece de la imunoglobulina normală umană la un alt preparat sau după un interval mai lung de la momentul perfuziei anterioare. Complicațiile potențiale pot fi evitate asigurându-se că pacienții: nu sunt hipersensibili la imunoglobulina normală umană prin administrarea inițială a preparatului într-un ritm lent (0,01 până la 0,02 ml / kg greutate corporală / min); sunt atent monitorizate pentru orice simptom pe parcursul perioadei de perfuzie.În special, pacienții care nu au primit anterior tratament cu imunoglobulină normală umană, pacienții care au primit anterior un preparat alternativ de IgIg sau dacă a trecut mult timp de la perfuzia anterioară trebuie monitorizați în timpul primei perfuzii și în prima oră după finalizarea acesteia. Toți ceilalți pacienți trebuie urmăriți timp de cel puțin 20 de minute. după administrarea preparatului. Dacă apare o reacție adversă, viteza de perfuzare trebuie redusă sau întreruptă. În caz de șoc, trebuie inițiat un tratament medical adecvat. La toți pacienții, administrarea intravenoasă de Ig necesită: o hidratare adecvată a pacientului înainte de începerea perfuziei de Ig intravenoasă, monitorizarea diurezei, monitorizarea creatininei serice, evitarea utilizării concomitente a diureticelor de ansă. Reacțiile de hipersensibilitate adevărate sunt rare: pot apărea la pacienții care au anticorpi împotriva IgA. Medicamentul nu este indicat la pacienții cu deficit selectiv de IgA la care deficitul de IgA este singura anomalie. În cazuri rare, administrarea de imunoglobulină umană poate determina scăderea tensiunii arteriale cu reacție anafilactică, chiar și la acei pacienți care au tolerat anterior tratamentul cu imunoglobulină umană. Infuziile cu IVIg trebuie administrate cu precauție la pacienții supraponderali și la pacienții cu factori de risc existenți pentru evenimente trombotice precum bătrânețe, hipertensiune arterială, diabet, boli vasculare sau antecedente de evenimente trombotice, tulburări trombotice dobândite sau congenitale, imobilizare prelungită, severă hipovolemie, boli în cursul cărora crește vâscozitatea sângelui. Au fost raportate cazuri de insuficiență renală acută la pacienții tratați cu preparate IVIg, factori de risc precum insuficiență renală preexistentă, diabet zaharat, hipovolemie, supraponderalitate, medicamente nefrotoxice concomitente, vârsta peste 65 de ani a fost identificată în majoritatea cazurilor. În cazurile de disfuncție renală, trebuie luată în considerare întreruperea tratamentului cu IgIV. Rapoartele despre disfuncție și insuficiență renală acută au fost asociate cu utilizarea multor produse IVIg aprobate (care conțin zaharoză, glucoză, maltoză ca excipienți), dar preparatele care conțin zaharoză ca stabilizator au contribuit în mod disproporționat la numărul total de cazuri; la pacienții cu risc, poate fi luată în considerare utilizarea produselor IVIg care nu conțin acești excipienți. La pacienții cu risc crescut de insuficiență renală acută sau reacții adverse tromboembolice, preparatele IVIg trebuie administrate la viteza minimă de perfuzie și doza posibilă. S-a raportat sindromul de meningită aseptică (AMS) în asociere cu terapia cu IgIV; întreruperea tratamentului cu IgIV a dus la remisiunea SMA în câteva zile fără sechele. Beneficiarii IVIg trebuie monitorizați pentru semne clinice și simptome de hemoliză. În ciuda măsurilor standard pentru prevenirea infecțiilor transmise prin preparate pe bază de sânge sau plasmă umană, atunci când se administrează astfel de preparate, posibilitatea de transmitere a agenților infecțioși nu poate fi exclusă în totalitate. Acest lucru se aplică și virușilor necunoscuți sau emergenți sau altor agenți patogeni. Metodele utilizate sunt considerate eficiente în prevenirea transmiterii virusurilor învelite, cum ar fi HIV, HBV și HCV, dar pot fi de utilizare limitată împotriva virusurilor neînvelite, cum ar fi HAV și parvovirusul B19. Experiența clinică arată că niciun virus al hepatitei A sau parvovirus B19 nu este transmis de preparatele de imunoglobulină. De asemenea, se presupune că prezența anticorpilor conținuți în aceste preparate contribuie la siguranța acestor preparate. Au fost raportate edeme pulmonare non-cardiogene (leziuni pulmonare acute post-transfuzionale - detresă respiratorie severă, edem pulmonar, hipoxemie, funcție ventriculară stângă normală și febră, 1-6 ore după transfuzie) la pacienții tratați cu IVIG, trebuie să se acorde atenție la administrarea Octagam deși până acum nu s-a observat un astfel de caz după administrarea acestui preparat. În timpul administrării IVIG poate apărea supraîncărcare cardiovasculară (volum), când volumul preparatului și al altor perfuzii administrate determină hipervolemie acută și edem pulmonar acut. Acest medicament conține nu mai mult de 0,03 mmol (0,69 mg) sodiu per ml de soluție. Acest lucru trebuie luat în considerare la pacienții care urmează o dietă controlată cu sodiu.
Activitate nedorită
Frecvente: hipersensibilitate, cefalee, greață, febră, oboseală, reacții la locul injectării. Mai puțin frecvente: eczeme, dureri de spate, frisoane, dureri în piept. Foarte rare: leucopenie, anemie hemolitică, șoc anafilactic, reacție anafilactică, reacție anafilactoidă, angioedem, edem facial, stare confuzională, neliniște, anxietate, infarct cerebral, meningită aseptică, pierderea cunoștinței, tulburări de vorbire, migrenă, amețeli, hipoestezie, parestezie, fotofobie, tremor, insuficiență vizuală, infarct miocardic, angina pectorală, bradicardie, tahicardie, palpitații, cianoză, tromboză, colaps circulator, insuficiență circulatorie periferică, flebită, hipotensiune arterială, hipertensiune, paloare, insuficiență respiratorie, , edem pulmonar, bronhospasm, hipoxie, dispnee, tuse, vărsături, diaree, dureri abdominale, descuamare a pielii, urticarie, erupție cutanată, erupție cutanată eritematoasă, dermatită, prurit, alopecie, eritem artralgic, durere musculară, durere la nivelul extremității, durere de gât , spasme musculare, slăbiciune musculară, rigiditate musculo-scheletică, insuficiență renală acută, durere de rinichi k, edem, simptome asemănătoare gripei, bufeuri, roșeață, senzație de frig, senzație de căldură, transpirație, stare generală de rău, disconfort toracic, astenie, letargie, senzație de arsură, enzime hepatice crescute, test de glicemie fals pozitiv.
Sarcina și alăptarea
Sarcina. Siguranța preparatului la femeile gravide nu a fost stabilită în studiile clinice controlate. Preparatul trebuie administrat femeilor însărcinate și femeilor care alăptează cu grijă deosebită. Preparatele IVIg traversează placenta, care crește în al treilea trimestru. Experiența clinică cu utilizarea imunoglobulinelor indică faptul că acestea nu au efecte dăunătoare asupra evoluției sarcinii, dezvoltării fătului sau a nou-născutului. Alăptarea. Imunoglobulinele sunt excretate în laptele matern și pot ajuta la protejarea nou-născutului de agenții patogeni care pătrund în mucoasă. Fertilitate. Experiența clinică cu utilizarea imunoglobulinelor sugerează că nu sunt de așteptat efecte nocive asupra fertilității.
Comentarii
Anumite reacții adverse pot afecta capacitatea de a conduce vehicule sau de a folosi utilaje. Pacienții care prezintă reacții adverse în timpul tratamentului trebuie să aștepte ca acestea să se rezolve înainte de a conduce vehicule sau de a folosi utilaje. După administrarea imunoglobulinei, poate exista o creștere tranzitorie a nivelului diferiților anticorpi transferați pasiv în sângele pacientului, ceea ce poate duce la rezultate fals pozitive în testele serologice. Anticorpii transferați pasiv împotriva antigenelor eritrocitare, de exemplu A, B, D, pot interfera cu rezultatele testelor serologice, de exemplu testul antiglobulinei directe (testul Coombs). La pacienții tratați cu preparate IVIG, rata de sedimentare a eritrocitelor (VSH) poate fi fals crescută (creștere neinflamatorie). Preparatul conține maltoză și, prin urmare, determinarea concentrației de glucoză trebuie efectuată folosind metode specifice glucozei.
Interacțiuni
Imunoglobulinele conținute în preparat pot reduce, în perioada de la 6 săptămâni la 3 luni, eficacitatea vaccinurilor vii, atenuate împotriva rujeolei, oreionului, rubeolei sau a varicelei (trebuie menținut un interval de 3 luni între administrare și vaccinare; în cazul vaccinării împotriva rujeolei, slăbiciune) Răspunsul imunitar poate persista până la un an, de aceea se recomandă măsurarea anticorpilor rujeolici înainte de vaccinare). Preparatul nu trebuie amestecat cu alte medicamente.
Preparatul conține substanța: imunoglobulină umană normală
Medicament rambursat: NU










---przyczyny-objawy-i-leczenie.jpg)