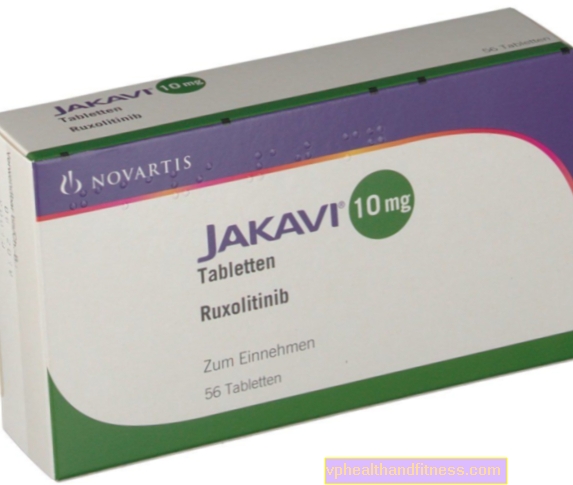
1 comprimat conține 5 mg, 10 mg, 15 mg sau 20 mg ruxolitinib sub formă de fosfat. Preparatul conține lactoză.
| Nume | Conținutul pachetului | Substanța activă | Preț 100% | Modificat ultima dată |
| Jakavi | 56 buc, masă | Ruxolitinib | 2019-04-05 |
Acțiune
Un medicament anticancer, un inhibitor al protein kinazei. Ruxolitinib este un inhibitor selectiv al kinazelor Janus (JAK), JAK1 și JAK2, care mediază semnalizarea pentru o serie de citokine și factori de creștere care joacă un rol important în hemopoieză și funcția imunitară. Se caracterizează prin permeabilitate ridicată, solubilitate bună și eliberare rapidă. Se absoarbe rapid după administrare orală, Cmax este atins la aproximativ 1 oră după administrare. Legarea de proteinele plasmatice este de aproximativ 97%, în principal cu albumina. Ruxolitinib nu traversează bariera hematoencefalică. Este metabolizat în principal de CYP3A4, cu contribuție suplimentară de la CYP2C9. În plasmă, medicamentul este prezent în principal sub formă nemodificată și ca doi metaboliți activi. Ruxolitinibul este eliminat în principal prin metabolism. Eliminarea medie T0.5 a ruxolitinibului este de aproximativ 3 ore, este excretată în principal în urină și fecale.
Dozare
Oral. Tratamentul trebuie inițiat numai de un medic cu experiență în administrarea de medicamente anti-cancer. Înainte de începerea tratamentului, trebuie efectuată o hemoleucogramă completă cu un test de globule albe. O hemogramă completă cu frotiu de globule albe trebuie efectuată la fiecare 2 - 4 săptămâni până când doza este stabilizată, apoi în funcție de indicațiile clinice. Doza inițială. Mielofibroză: 15 mg de două ori pe zi la pacienții cu număr de trombocite între 100.000 / mm3 și 200.000 / mm3 și 20 mg de două ori pe zi la pacienții cu număr de trombocite> 200.000 / mm3. Policiteemia Vera: 10 mg pe cale orală de 2 ori pe zi. Există informații limitate cu privire la doza inițială recomandată pentru pacienții cu număr de trombocite între 50.000 / mm3 și 3 - max. doza inițială recomandată la acești pacienți este de 5 mg de două ori pe zi și trebuie crescută cu precauție. Modificări ale dozelor. Dozele pot fi ajustate pe baza siguranței și eficacității medicamentului. Tratamentul trebuie întrerupt dacă numărul de trombocite este mai mic de 50.000 / mm3 sau numărul absolut de neutrofile este mai mic de 500 / mm3. Tratamentul trebuie oprit și la pacienții cu PV dacă nivelul hemoglobinei este sub 8 g / dL. După creșterea numărului de sânge peste aceste valori, dozarea poate fi reluată la o doză de 5 mg de două ori pe zi, cu o creștere treptată pe baza rezultatelor unui test de sânge complet cu frotiu. Trebuie luată în considerare reducerea dozei dacă numărul de trombocite scade sub 100.000 / mm3 pentru a evita întreruperea tratamentului din cauza trombocitopeniei. Reducerea dozei trebuie luată în considerare și la pacienții cu PV dacă hemoglobina este sub 12 g / dl și se recomandă reducerea dozei dacă hemoglobina este sub 10 g / dL. Dacă tratamentul este considerat insuficient de eficient și hemoleucograma este adecvată, doza poate fi crescută cu maxim 5 mg de două ori pe zi, până la o valoare maximă. doze de 25 mg de două ori pe zi. Doza inițială nu trebuie crescută în primele patru săptămâni de tratament și, ulterior, nu trebuie crescută mai frecvent decât la intervale de 2 săptămâni. Max. doza preparatului este de 25 mg de două ori pe zi. Ajustările dozei atunci când se iau concomitent inhibitori puternici ai CYP3A4 sau fluconazol. O doză unitară trebuie redusă cu aproximativ 50% și administrată de două ori pe zi. Trebuie evitată utilizarea simultană a preparatului cu fluconazol în doze mai mari de 200 mg pe zi. În timpul tratamentului cu inhibitori puternici ai CYP3A4 sau cu inhibitori duali ai enzimelor CYP2C9 și CYP3A4, se recomandă monitorizarea mai frecventă (de exemplu de două ori pe săptămână) a parametrilor hematologici și a semnelor și simptomelor reacțiilor adverse legate de medicament. Întreruperea tratamentului. Tratamentul trebuie continuat atâta timp cât raportul beneficiu-risc rămâne pozitiv, dar trebuie întrerupt după 6 luni dacă nu a existat nicio reducere a dimensiunii splinei sau îmbunătățirea simptomelor de la începerea tratamentului. Se recomandă ca pacienții care prezintă un anumit grad de ameliorare clinică să întrerupă ruxolitinibul dacă prezintă o alungire a splinei de 40% comparativ cu lungimea inițială (aproximativ echivalentă cu o creștere a volumului splinei de 25%) și nu se observă nicio îmbunătățire reală. în raport cu simptomele asociate bolii. Grupuri speciale de pacienți. Nu este necesară ajustarea dozei speciale la pacienții cu insuficiență renală ușoară sau moderată. La pacienții cu insuficiență renală severă (CCr 3 până la 200.000 / mm3. Se recomandă o doză unică de 20 mg sau 2 doze de 10 mg administrate la 12 ore distanță la pacienții cu MF cu număr de trombocite> 200.000 / mm3. Dozele ulterioare (administrare unică sau 2 doze de 10 mg la 12 ore distanță) trebuie administrate numai în zilele de hemodializă, după fiecare ședință de dializă. Doza inițială recomandată pentru pacienții cu hemodializă cu ESRD și PV este o doză unică de 10 mg sau două doze după fiecare ședință de dializă. 5 mg administrate la intervale de 12 ore după dializă și numai în ziua de hemodializă Aceste recomandări de dozare sunt simulate și orice modificare a dozei la pacienții cu ESRD trebuie monitorizată cu atenție pentru siguranță și eficacitate. Nu există date disponibile la pacienții care urmează tratament. dializă peritoneală sau hemofiltrare veno-venoasă continuă La pacienții cu orice funcție hepatică afectată, doza inițială recomandată se bazează pe și o trombocite trebuie reduse cu aproximativ 50% și administrate de două ori pe zi. Dozele ulterioare trebuie ajustate pe baza siguranței și eficacității medicamentului. Pacienții diagnosticați cu disfuncție hepatică în timpul tratamentului cu preparatul ar trebui să facă un test de sânge complet cu frotiu cel puțin 1 din 1-2 săptămâni în primele 6 săptămâni după începerea tratamentului și apoi, după stabilizarea funcției hepatice și teste de sânge - dacă sunt disponibile. indicații clinice. Doza poate fi ajustată pentru a reduce riscul de citopenie. Nu sunt recomandate ajustări suplimentare ale dozei la vârstnici. Siguranța și eficacitatea la copiii cu vârsta sub 18 ani nu au fost stabilite. Nu există date disponibile. Mod de a da. Luați cu sau fără alimente. Dacă se omite o doză, pacientul nu trebuie să ia o doză suplimentară, ci să ia următoarea doză prescrisă.
Indicații
Fibroza maduvei. Tratamentul splenomegaliei asociate cu o boală sau simptome observate la pacienții adulți cu fibroză a măduvei osoase primare (cunoscută și sub numele de fibroză idiopatică cronică a măduvei osoase), fibroză a măduvei osoase precedată de policitemie (hiperaemie) vera sau fibroză a măduvei osoase precedată de trombocitemie esențială. Rată tuftată. Tratamentul pacienților adulți cu policitemie vera care sunt rezistenți sau intoleranți la terapia cu hidroxicarbamidă.
Contraindicații
Hipersensibilitate la substanța activă sau la oricare dintre excipienți. Sarcina și alăptarea.
Precauții
Medicamentul poate provoca reacții adverse hematologice, inclusiv trombocitopenie, anemie și neutropenie, de aceea este necesară o analiză completă a sângelui cu frotiu de globule albe înainte de începerea tratamentului. Tratamentul trebuie întrerupt la pacienții cu un număr de trombocite mai mic de 50.000 / mm3 sau cu un număr absolut de neutrofile mai mic de 500 / mm3. S-a observat că pacienții cu număr scăzut de trombocite (3) la inițierea terapiei sunt mai predispuși să dezvolte trombocitopenie în timpul tratamentului. Trombocitopenia este în general reversibilă și poate fi gestionată de obicei prin reducerea dozei sau reținerea temporară a preparatului, deși transfuziile de trombocite pot fi necesare în funcție de indicația clinică. Pacienții care dezvoltă anemie pot necesita transfuzii de sânge, iar ajustarea dozei sau întreruperea tratamentului poate fi luată în considerare la pacienții cu anemie. Pacienții cu niveluri de hemoglobină sub 10,0 g / dL la începutul tratamentului prezintă un risc mai mare de niveluri de hemoglobină sub 8,0 g / dL în timpul tratamentului comparativ cu pacienții cu niveluri de hemoglobină inițiale mai ridicate, prin urmare la pacienții cu niveluri inițiale de hemoglobină. hemoglobină sub 10,0 g / dl, se recomandă monitorizarea mai frecventă a parametrilor hematologici, precum și evaluarea semnelor și simptomelor care indică efectele adverse asociate cu utilizarea preparatului. Neutropenia (numărul absolut de neutrofile <500 / mm3) a fost în general reversibilă și a fost gestionată prin deținerea temporară a medicamentului. Un test complet de sânge trebuie efectuat de câte ori este indicat clinic și ajustarea dozei, după cum este necesar. La pacienții tratați cu preparatul au apărut infecții grave bacteriene, micobacteriene, fungice, virale și alte oportuniste, prin urmare, pacienții trebuie evaluați pentru riscul de infecții grave. Medicii trebuie să monitorizeze îndeaproape pacienții care primesc medicamentul pentru semne și simptome ale infecțiilor și să instituie imediat un tratament adecvat. Tratamentul nu trebuie început până când nu mai există o infecție activă gravă. Tuberculoza a fost raportată la pacienții care iau medicamentul pentru mielofibroză, iar pacienții trebuie testați pentru tuberculoză activă sau inactivă (latentă) înainte de a începe tratamentul, conform recomandărilor locale. Investigațiile ar trebui să includă antecedente medicale, posibile contacte anterioare cu pacienți cu tuberculoză și / sau teste de screening adecvate, cum ar fi radiografia pulmonară, testul tuberculinei și / sau, dacă este cazul, testul de eliberare a interferonului-γ. Medicii trebuie să fie conștienți de riscul unui test cutanat fals negativ la tuberculină, în special la pacienții grav bolnavi sau imunocompromiși. Au fost raportate creșteri ale virusului hepatitei B (titru VHB-ADN), cu sau fără creșteri concomitente ale ALT și AST la pacienții cu infecție cronică VHB care iau acest medicament. Efectul medicamentului asupra replicării virale la pacienții cu infecție cronică cu VHB este necunoscut; pacienții cu VHB cronici trebuie tratați și monitorizați conform ghidurilor clinice. Din cauza riscului de zona zoster, medicii trebuie să instruiască pacienții să recunoască semnele și simptomele timpurii, recomandând începerea tratamentului cât mai curând posibil. Leukoencefalopatia multifocală progresivă (LMP) a fost raportată cu utilizarea Jakavi pentru tratamentul MF, prin urmare, medicii ar trebui să fie vigilenți cu privire la simptomele care pot sugera LMP pe care pacienții nu le pot observa (de exemplu, cognitive, neurologice sau mental). Pacienții trebuie monitorizați pentru simptome noi sau agravante și, în cazul în care apar astfel de simptome, trebuie avută în vedere trimiterea pacientului la un neurolog sau instituirea unor măsuri de diagnostic adecvate. Dacă se suspectează LMP, tratamentul suplimentar trebuie suspendat până la excluderea LMP. Neoplasmele maligne ale pielii non-melanom (NMSC) (inclusiv carcinom bazocelular, carcinom scuamos și carcinom celular Merkel) au fost raportate la pacienții tratați cu ruxolitinib, majoritatea acestor pacienți cu antecedente de terapie pe termen lung cu hidroxicarbamidă și anterior NMSC sau leziuni cutanate precanceroase. Se recomandă examinarea periodică a pielii la pacienții cu risc crescut de cancer de piele. Tratamentul cu preparatul a fost asociat cu creșteri ale parametrilor lipidici, incluzând colesterolul total, colesterolul HDL, colesterolul LDL și trigliceridele - se recomandă monitorizarea nivelurilor de lipide și tratarea dislipidemiei conform ghidurilor clinice. Doza inițială trebuie redusă la pacienții cu insuficiență renală severă. La pacienții cu boală renală în stadiu final și MF care primesc hemodializă, doza inițială trebuie să se bazeze pe numărul de trombocite; dozele ulterioare trebuie administrate numai în ziua de hemodializă după sfârșitul fiecărei ședințe de hemodializă. Ar trebui efectuate ajustări suplimentare ale dozei cu o monitorizare atentă a siguranței și eficacității medicamentului. Doza inițială trebuie redusă cu aproximativ 50% la pacienții cu insuficiență renală. Alte ajustări ale dozelor trebuie făcute pe baza siguranței și eficacității medicamentelor. Dacă preparatul trebuie administrat concomitent cu inhibitori puternici ai CYP3A4 sau cu inhibitori duali ai enzimelor CYP3A4 și CYP2C9 (de exemplu, fluconazol), doza unitară trebuie redusă cu aproximativ 50% și administrată de două ori pe zi. Utilizarea concomitentă a medicamentelor cu factor de creștere citoreductiv sau hematopoietic și preparatul nu a fost studiată. După întreruperea sau întreruperea tratamentului, simptomele MF pot reveni în aproximativ o săptămână. Există cazuri cunoscute de pacienți care au întrerupt tratamentul cu preparatul care se confruntă cu evenimente mai severe, în special cei cu alte boli comorbide acute. Nu se știe dacă întreruperea bruscă a tratamentului a contribuit la apariția acestor evenimente. Reducerea treptată a dozei poate fi luată în considerare, cu excepția cazului în care este necesară întreruperea bruscă a tratamentului, deși utilitatea reducerii dozei nu a fost dovedită. Preparatul conține lactoză - nu trebuie utilizat la pacienții cu probleme ereditare rare de intoleranță la galactoză, deficit de lactază Lapp sau malabsorbție de glucoză-galactoză.
Activitate nedorită
La pacienții cu policitemie vera. Foarte frecvente: infecții ale tractului urinar, anemie CTCAE de gradul 3 (3) și gradul 3 (50.000 - 25.000 / mm3), neutropenie de gradul 3.(3) și 4 (3) CTCAE, hemoragie intracraniană, sângerare gastrointestinală, alte sângerări (inclusiv epistaxis, hemoragii postoperatorii și hematurie), flatulență, creșterea CTCAE grad 3 alanină aminotransferază ( > 5x - 20x ULN). Mai puțin frecvente: tuberculoză. La pacienții cu mielofibroză. Foarte frecvente: grad CTCAE orice anemie, grad CTCAE orice trombocitopenie, sângerare (orice sângerare incluzând sângerări intracraniene și gastrointestinale, vânătăi și alte sângerări, vânătăi, alte sângerări (inclusiv epistaxis, post-procedural și Hemataturie), hipercolesterolemie CTCAE de gradul 1 și 2, hipertrigliceridemie CTCAE de gradul 1, amețeli, creșteri ale alaninei și aspartatului aminotransferazei CTCAE de grad 1, hipertensiune arterială Frecvente: infecții ale tractului urinar, zona zoster, trombocitopenie de gradul 3 (50 CTCAE 000 - 25.000 / mm3), creștere în greutate, constipație Mai puțin frecvente: anemia CTCAE de gradul 3 (3) alanină aminotransferază crescută (> 5x - 20x LSN) după întreruperea tratamentului la pacienți Cu MF, poate apărea reapariția simptomelor MF, cum ar fi oboseală, dureri osoase, febră, mâncărime, transpirații nocturne, mărirea simptomatică a splinei și pierderea în greutate. În studiile clinice cu MF, scorul total al simptomului MF a revenit treptat la valoarea inițială în termen de 7 zile de la întreruperea tratamentului.
Sarcina și alăptarea
Utilizarea preparatului în timpul sarcinii este contraindicată. Femeile aflate la vârsta fertilă ar trebui să utilizeze metode contraceptive eficiente în timpul tratamentului și, dacă o femeie rămâne gravidă în timpul tratamentului cu preparatul, ar trebui făcută o evaluare individuală a raportului beneficiu-risc cu consiliere cu privire la posibilul risc pentru făt. Nu trebuie utilizat în timpul alăptării și, prin urmare, alăptarea trebuie întreruptă la începerea tratamentului.
Comentarii
Nu are niciun efect sedativ sau neglijabil. Cu toate acestea, pacienții care au amețeli după ce au luat preparatul ar trebui să se abțină de la conducerea vehiculelor sau de la utilizarea utilajelor.
Interacțiuni
Studiile de interacțiune au fost efectuate numai la adulți. Ruxolitinib este eliminat prin metabolism catalizat de CYP3A4 și CYP2C9, prin urmare, preparatele care inhibă activitatea acestor enzime pot duce la expunerea crescută la ruxolitinib. Când se administrează medicamentul cu inhibitori puternici ai CYP3A4 (cum ar fi, dar fără a se limita la, iboceprevir, claritromicină, indinavir, itraconazol, ketoconazol, lopinavir / ritonavir, ritonavir, mibefradil, nefazodonă, nelfinavir, posaconazol, un univeravir) 50% și administrat de două ori pe zi. Pacienții trebuie monitorizați îndeaproape (de exemplu, de două ori pe săptămână) pentru posibila citopenie, iar doza trebuie crescută treptat pe baza siguranței și eficacității. Trebuie luată în considerare o reducere a dozei de 50% atunci când se utilizează preparate care sunt inhibitori duali ai enzimelor CYP2C9 și CYP3A4 (de exemplu, fluconazol). Trebuie evitată utilizarea concomitentă a medicamentului cu fluconazol în doze mai mari de 200 mg pe zi. Atunci când se iau inductori ai CYP3A4 (cum ar fi, dar fără a se limita la avasimibe, carbamazepină, fenobarbital, fenitoină, rifabutină, rifampicină (rifampicină), sunătoare (Hypericum perforatum), pacienții trebuie monitorizați îndeaproape și doza trebuie crescută treptat pe baza siguranței și eficacității. Nu se recomandă ajustarea dozei atunci când ruxolitinib se administrează concomitent cu inhibitori ușori sau moderate ai CYP3A4 (cum ar fi, dar fără a se limita la, ciprofloxacină, eritromicină, amprenavir, atazanavir, diltiazem, cimetidină), cu toate acestea, pacienții trebuie monitorizați îndeaproape pentru posibila citopenie la inițierea terapiei cu inhibitori moderate. CYP3A4: Ruxolitinibul poate inhiba glicoproteina intestinală P și proteina de rezistență la cancerul de sân (BCRP), ceea ce ar putea duce la o expunere sistemică crescută a substraturilor acestor transportori precum dabigatran etexilat, ciclosporină, rosuvastatină și potențial digoxină. monitorizarea medicamentelor (TDM) sau starea clinică după administrarea substanțelor menționate. Este posibil ca inhibarea potențială a P-gp și BCRP în intestin să fie redusă la minimum dacă timpul dintre administrările medicamentului este menținut cât mai mult posibil. Utilizarea concomitentă a factorilor de creștere hematopoietici și a preparatului nu a fost studiată. Nu se știe dacă inhibarea kinazelor Janus (JAK) de către Jakavi reduce eficacitatea factorilor de creștere hematopoietici sau dacă factorii de creștere hematopoietici afectează eficacitatea preparatului. Utilizarea simultană a terapiilor citoreductive și preparatul nu au fost studiate - siguranța și eficacitatea administrării simultane a acestor medicamente este necunoscută. Ruxolitinib nu inhibă metabolismul midazolamului substratului CYP3A4 oral - prin urmare, nu este de așteptat o creștere a expunerii la substraturile CYP3A4 atunci când aceste medicamente sunt administrate concomitent cu Jakavi. Preparatul nu afectează farmacocinetica unui contraceptiv oral care conține etinilestradiol și levonorgestrel, prin urmare eficacitatea contraceptivelor care conțin această combinație nu este de așteptat să fie redusă în timpul utilizării concomitente a ruxolitinib.
Preparatul conține substanța: Ruxolitinib
Medicament rambursat: NU











---przyczyny-objawy-i-leczenie.jpg)